5.1 EMP_collapse
Data collapse refers to merging the original assay to generate a new assay. EMP_collapse can work based on the specified data in rowdata or coldata. Users can calculate the average, median, sum, maximum, or minimum as the combined result during the folding process.
5.1.1 Merge assays based on duplicate coldata
EMP_collapse can collapse the assay according to the annotation in the coldata.
🏷️Example: Merge the assay data according to the column Group in the coldata.
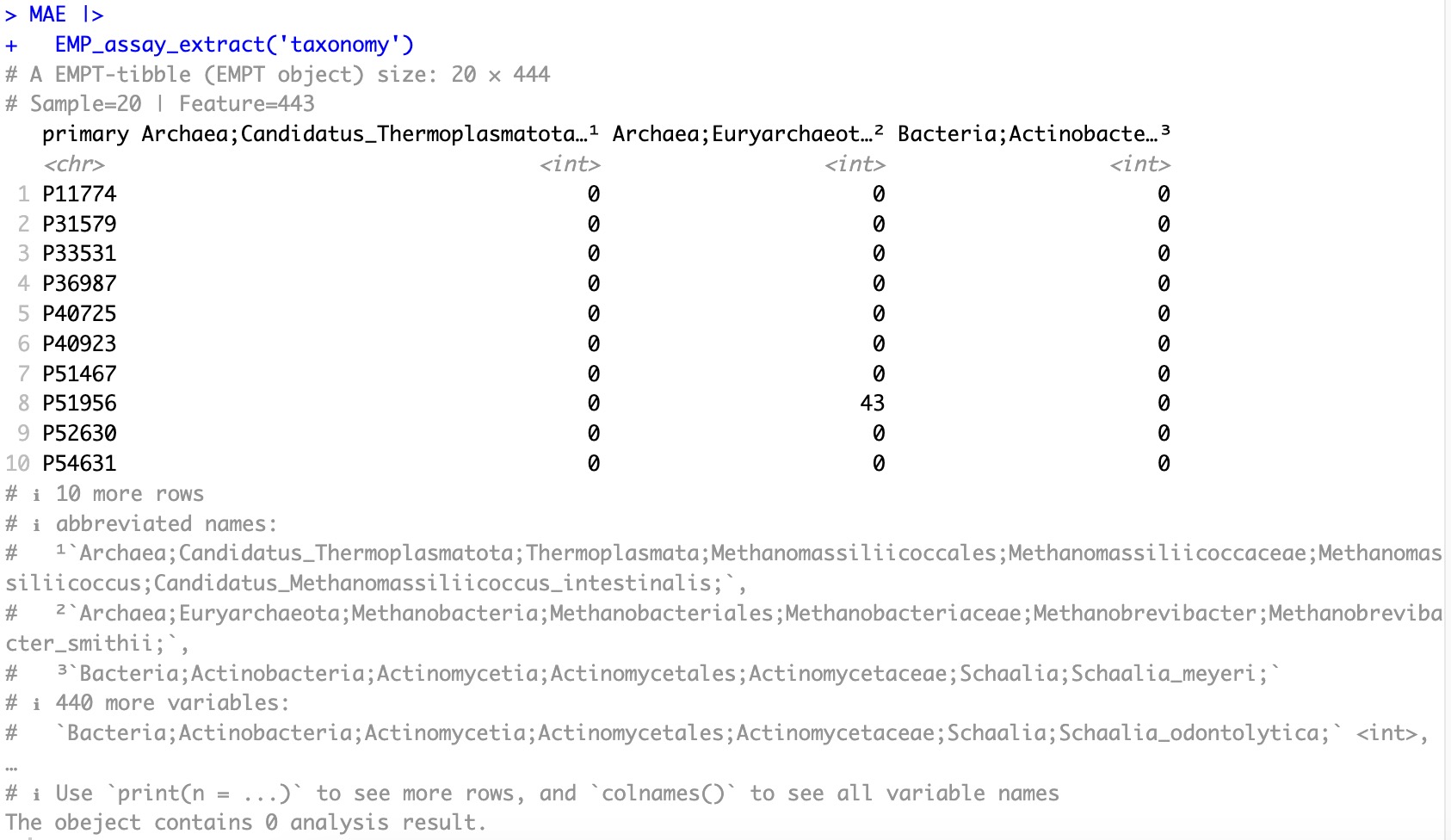
Original assay:
MAE |>
EMP_assay_extract(experiment='taxonomy')
Folded assay:
Extract the assay from taxonomy experiment. The parameter estimate_group specifies that the data should be aggregated based on the Group column in coldata. The parameter collapse_by indicates that aggregation should be performed according to the col column in coldata. The parameter collapse_sep specifies that the + delimiter should be used to concatenate the string cells in the coldata where aggregation occurs.
MAE |>
EMP_assay_extract(experiment='taxonomy') |>
EMP_collapse(collapse_by='col',estimate_group = 'Group',
method = 'mean',collapse_sep = '+')

Folded coldata:
Although the data aggregation operation essentially involves collapsing the
assay, the assay, coldata, and rowdata are intrinsically linked from the moment the MAE object is created. Therefore, during the collapse process, the coldata is also aggregated accordingly. For continuous variables in coldata, the aggregation method used will be the same as that for the assay. For categorical variables in coldata, the specified collapse_sep delimiter will be used to concatenate the data. For boolean/logical variables, the parameter collapse_boolean will be used to concatenate the data.
MAE |>
EMP_assay_extract(experiment='taxonomy') |>
EMP_collapse(collapse_by='col',estimate_group = 'Group',
method = 'mean',collapse_sep = '+') |>
EMP_coldata_extract()
5.1.2 Merge assays based on duplicate rowdata
EMP_collapse can collapse the assay according to the annotation in the rowdata.
🏷️Example 1: Merge the assay data according to the column Class in the rowdata.
In microbiome studies, the EMP_collapse function can rapidly aggregate the features of the assay from higher taxonomic levels (such as Phylum or Genus) to lower levels by specifying the estimate_group parameter.
①After collapsing the assay based on rowdata, the feature of the assay will change, but the sample remains still. In this example, features originally at the species level are aggregated to the Class level.
②During the microbial data in this process, conflicts may arise between complete-level annotation and single-level annotation. Please refer to the Microbial Data Full Annotation Issue for more details.
③In this process, this module will automatically detect the aforementioned conflicts and prompt the user to choose between complete-level annotation and single-level annotation. Since the module defaults to automatic caching during the process, if you need to make a new selection, you can set
use_cached = FALSE and then re-run the process. MAE |>
EMP_assay_extract(experiment='taxonomy') |>
EMP_collapse(collapse_by='row',estimate_group = 'Class',
method = 'sum',collapse_sep = '+')

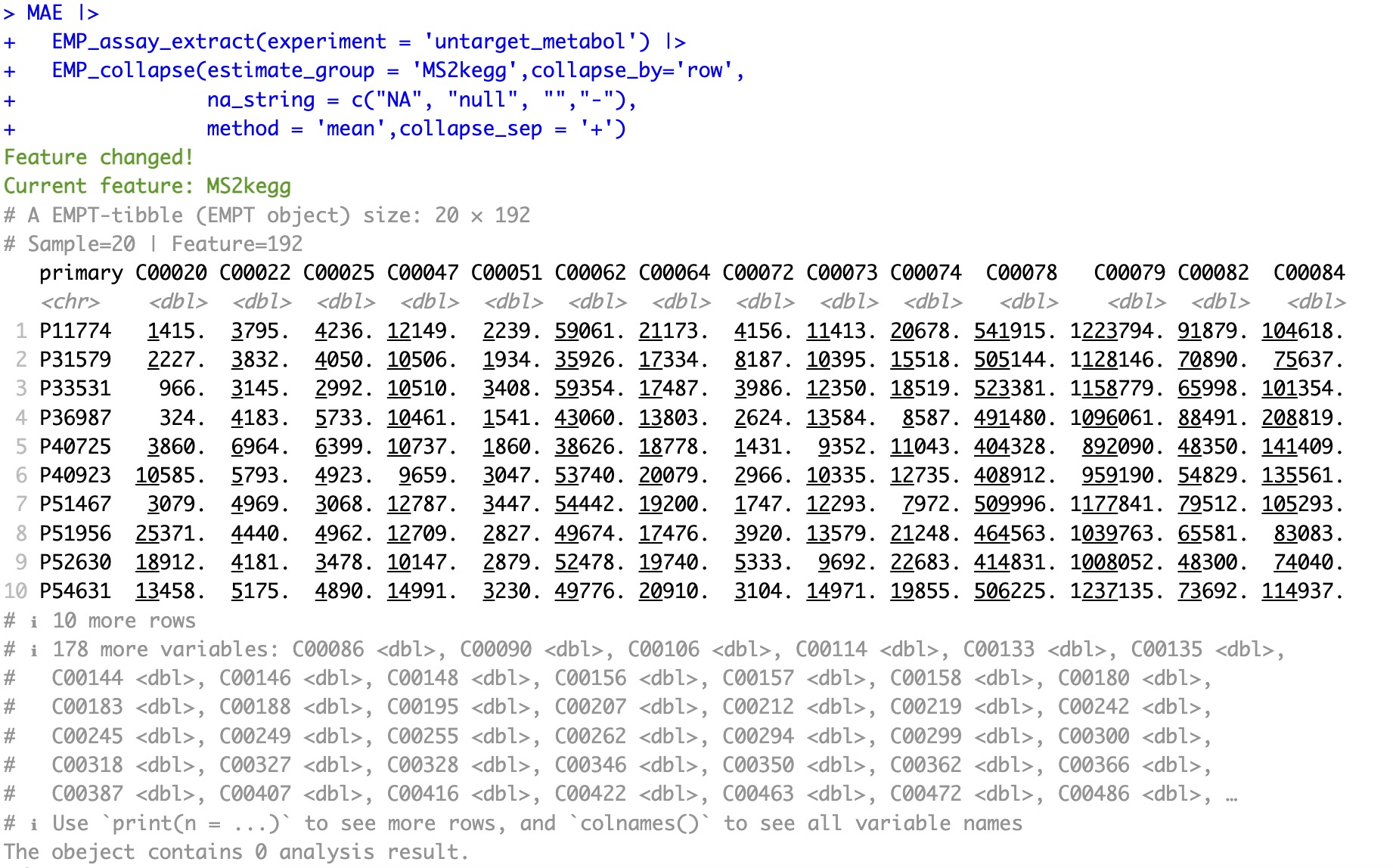
🏷️Example 2: Merge the assay metabolite data according to rowdata.
①In metabolomics projects, metabolites often have multiple annotation levels. Users can quickly aggregate the assay data according to the annotation level of metabolites in rowdata.
②The features used for aggregation must be present in rowdata (for example, the assay in the geno_ko project cannot be collapsed according to the Class column in rowdata). Users can use the function
EMP_rowdata_extract to view the feature annotation available for aggregation.
Original assay:
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol')

Folded assay: Merge the assay data according to the column MS2kegg in the rowdata.
In some metabolomics raw data, symbols such as
NA, null, and - are commonly used to represent missing values. The parameter na_string allows users to specify these strings as missing values. During data aggregation, any rows containing missing values in the specified features for merging will be ignored.
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol') |>
EMP_collapse(estimate_group = 'MS2kegg',collapse_by='row',
na_string = c("NA", "null", "","-"),
method = 'mean',collapse_sep = '+')